Public Data: TCGA Breast CancerWe are pleased to announce that data from the following paper have been made available on GenomeSpace: The GenomeSpace mirror of this dataset includes all public level 3 and level 4 data from https://tcga-data.nci.nih.gov/docs/publications/brca_2012/. You can access the data in GenomeSpace at the path /Public/SharedData/Datasets/TCGA/Breast Cancer/. (This requires a GenomeSpace login. If you don't have one, it's easy to register.) Abstract:We analyzed primary breast cancers by genomic DNA copy number arrays, DNA methylation, exome sequencing, mRNA arrays, microRNA sequencing, and reverse phase protein arrays. Our ability to integrate information across platforms provided key insights into previously-defined gene expression subtypes and demonstrated the existence of four main breast cancer classes when combining data from five platforms, each of which shows significant molecular heterogeneity. Somatic mutations in only three genes (TP53, PIK3CA and GATA3) occurred at > 10% incidence across all breast cancers; however, there were numerous subtype-associated and novel gene mutations including the enrichment of specific mutations in GATA3, PIK3CA, and MAP3K1 with the Luminal A subtype. We identified two novel protein expression-defined subgroups, possibly contributed by stromal/microenvironmental elements, and integrated analyses identified specific signaling pathways dominant in each molecular subtype including a HER2/p-HER2/HER1/p-HER1 signature within the HER2-Enriched expression subtype. Comparison of Basal-like breast tumors with high-grade Serous Ovarian tumors showed many molecular commonalities, suggesting a related etiology and similar therapeutic opportunities. The biologic finding of the four main breast cancer subtypes caused by different subsets of genetic and epigenetic abnormalities raises the hypothesis that much of the clinically observable plasticity and heterogeneity occurs within, and not across, these major biologic subtypes of breast cancer. GenomeSpace UI Changes Summary, November 2012Over the last several months there have been numerous changes and updates to GenomeSpace, not all of which received their own blog posts. Below is a summary of many of the more interesting recent changes. System Menu Renamed Manage
The System menu has been renamed Manage. New menu items have been added to the Manage menu to permit searching for a tool and mounting storage options external to GenomeSpace (currently your own or public Amazon S3 buckets with more options to come in the future). Tools and Data Sources Combined
Formerly, the analysis tools and datasources were kept in separate lists at the top of your page. We have changed GenomeSpace to keep these in a single list now and have added a slider to allow you to navigate between them when there are too many to fit on the screen. Drag-and-Drop Move of Multiple Files
If you select multiple files using the checkboxes, you may now drag-and-drop to move them all to a new directory. Previously, only the one file you were dragging would be moved, not all selected files. Launch Tools on External URLs
You can now drag a URL from your browser and drop it onto the dropzone in the tool launch dialog to launch a GenomeSpace tool on that URL. This allows you to launch GenomeSpace tools on URLs from any web site, not just GenomeSpace. Help Menu AdditionsWe have added links to the GenomeSpace support page and an email link (Contact us) to the Help menu to make it easier for you to get help with the GSUI or the GenomeSpace tools. Previous blog posts about GSUI Features
Public Data: TCGA Squamous Cell Lung CancerWe are pleased to announce that data from the following paper have been made available on GenomeSpace: The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. The GenomeSpace mirror of this dataset includes all public level 3 and level 4 data from https://tcga-data.nci.nih.gov/docs/publications/lusc_2012/. You can access the data in GenomeSpace at the path /Public/SharedData/Datasets/TCGA/Squamous Cell Lung Cancer/. (This requires a GenomeSpace login. If you don't have one, it's easy to register.) AbstractLung squamous cell carcinoma is a common type of lung cancer, causing approximately 400,000 deaths per year worldwide. Genomic alterations in squamous cell lung cancers have not been comprehensively characterized, and no molecularly targeted agents have been specifically developed for its treatment. As part of The Cancer Genome Atlas, we profile 178 lung squamous cell carcinomas to provide a comprehensive landscape of genomic and epigenomic alterations. We show that the tumour type is characterized by complex genomic alterations, with a mean of 360 exonic mutations, 165 genomic rearrangements, and 323 segments of copy number alteration per tumour. We find statistically recurrent mutations in 11 genes, including mutation of TP53 in nearly all specimens. Previously unreported loss-of-function mutations are seen in the HLA-A class I major histocompatibility gene. Significantly altered pathways included NFE2L2 and KEAP1 in 34%, squamous differentiation genes in 44%, phosphatidylinositol-3-OH kinase pathway genes in 47%, and CDKN2A and RB1 in 72% of tumours. We identified a potential therapeutic target in most tumours, offering new avenues of investigation for the treatment of squamous cell lung cancers. Prototype: Lightweight web site integration with GenomeSpaceThe GenomeSpace team has created a prototype interface to allow web applications to easily send files available at publicly accessible URLs to a GenomeSpace User's Home directory without the need to integrate with the CDK or otherwise directly use the API. To find out how to use this, please see the lightweight data source integration documentation. Please note that this is still a prototype and the exact details of this API may change in the future. New Feature: Tool tagging and search<The GenomeSpace team is pleased to announce the addition of tool tagging and search to GenomeSpace. These new features allow anyone in the GenomeSpace community to publicly or privately annotate tools to reflect their capabilities and use, and to search through ones own and public annotations to find tools with particular annotations, names, creators, or descriptions. Tagging a ToolTo tag a tool, you start by opening the tool's drop-down in the tool bar. Select Tags to view the current tags on a tool.
From this dialog box, you can add a new tag (optionally including a description) or remove a tag that you put onto a tool. You cannot remove tags placed by other users however. To add a tag in this dialog box:
To remove a tag from the tool:
Searching ToolsTo search through tool tags, names, or descriptions, select System>Search for Tools from the menu bar.
Enter your search term(s) and click Search Tools. All searches are case-insensitive (i.e., GENE will match to gene, Gene, geNe, etc). The search results display the tools with all their tags. The matching search term is bolded in the tool name/description and matching tags are shown and clicking on a tag in the search results will cause a new search to be made using that tag as the search term.
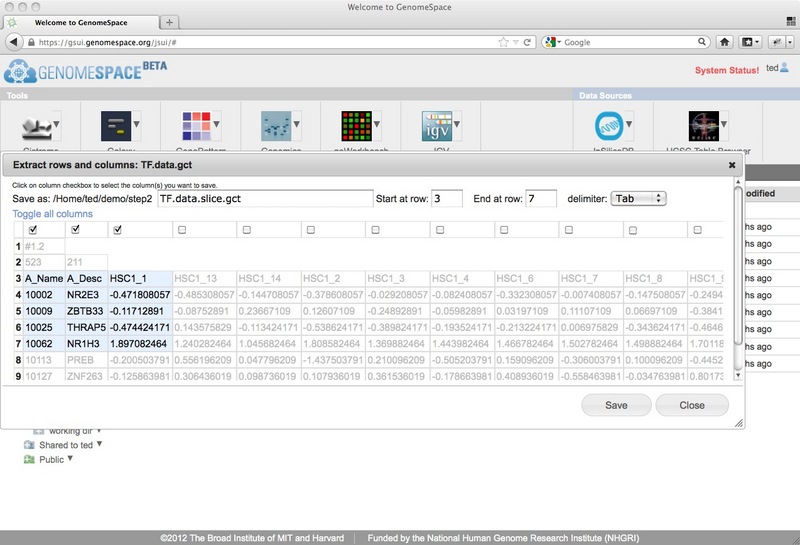
New Feature: Extracting rows and columns from a fileThe GenomeSpace team is pleased to announce a new GenomeSpace feature: the addition of server-side row and column extraction. GenomeSpace now has server-side row and column extraction that allows you to pull a set of rows and columns out of your tab- or comma-delimited data file and save it as its own file in GenomeSpace, without having to download the original file or re-upload the resulting filtered file. The Extract Rows and Columns dialog allows you to quickly and easily:
To access this feature you can either:
This opens a dialog, showing only the first 10 or so lines (or the first 50kb if there are a lot of columns) of the selected file.
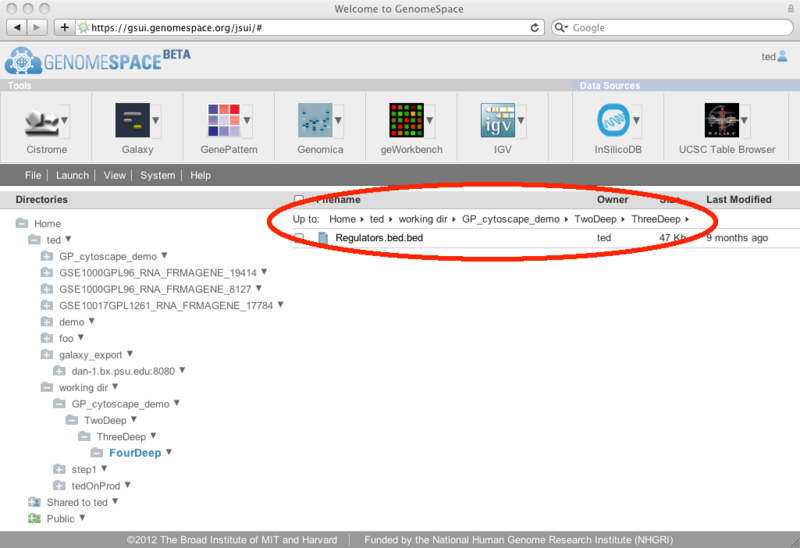
From this dialog, you can select the first row at which to start the row and column extraction (you can trim out header lines by starting at a lower row, for example) and the last row to include (leave this blank to take the rest of the file from the starting row). Then you can select columns by checking the checkbox at the top of each column. If you want to select many columns at once, you can use the 'toggle all columns' link which will check any unchecked columns and uncheck any that were previously checked. The rows/columns that have been selected to go to the new file are highlighted in light blue, while those that will be cut out are displayed as grey text on a white background. Finally, decide on the file name you want to use for the new subset file. The default will be to add .slice to the end of the source filename, leaving the file extension intact. For example, if your source filename is myfile.gct, the default extracted file name will be myfile.slice.gct. Note that if you are removing header lines you may also want to change the file extension to match the new format. Click Save to create a new GenomeSpace file in the same GenomeSpace directory as the original file. New Feature: Links to parent directoriesThe GenomeSpace team is pleased to announce a new GenomeSpace feature: the addition of parent directory links. We have added breadcrumb links to the top of the right pane of GenomeSpace to make it easier to navigate around directories in your cloud-based file system. Clicking on any of the links in the breadcrumbs will open that directory in GenomeSpace.
When you are at the top level of the directory tree (i.e., /Home), the links are not present since it is not possible for you to navigate any higher in the directory system.
New Tool: geWorkbenchGenomeSpace is proud to announce the addition of another new tool to the GenomeSpace tool bar: geWorkbench, the bioinformatics platform of Columbia University’s National Center for the Multiscale Analysis of Genomic and Cellular Networks. geWorkbench (genomics Workbench) is a Java-based open-source platform for integrated genomics. Using a component architecture, it allows individually developed plug-ins to be configured into complex bioinformatic applications. At present there are more than 70 available plug-ins supporting the visualization and analysis of gene expression and sequence data. You should see geWorkbench appear in your tool bar the next time you login to the GenomeSpace User Interface (GSUI) or refresh the page. Please note that since geWorkbench is a large application, the first time you launch it you will need to download geWorkbench. A link will be provided when you launch geWorkbench from GenomeSpace. Adding Private Tools: Part 2About a month ago, we posted that GenomeSpace had added the ability to let you put your own tools into the tool bar so that you could use your local instance of GenePattern, Galaxy, or any other GenomeSpace enabled tool. It has now gotten easier to add your tools into GenomeSpace with the release of the Manage Private Tools dialog. To add a tool to your toolbar, first, make sure it is GenomeSpace enabled. Once the tool is ready, start at the System menu and select Manage Private Tools.
This will open up the list of private tools that you have available. Most people will start with an empty list like this (below). Click Add new to start the process of adding a new tool.
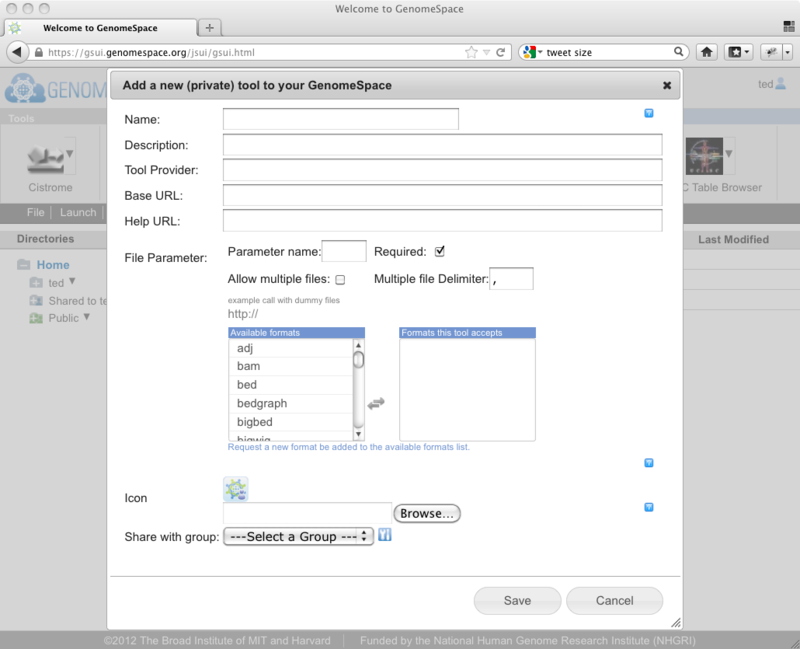
This will present you with the dialog for adding your tool to the tool bar.
As we discussed in the earlier post, you now need to enter the information about the tool that you are adding. Here you will need to enter:
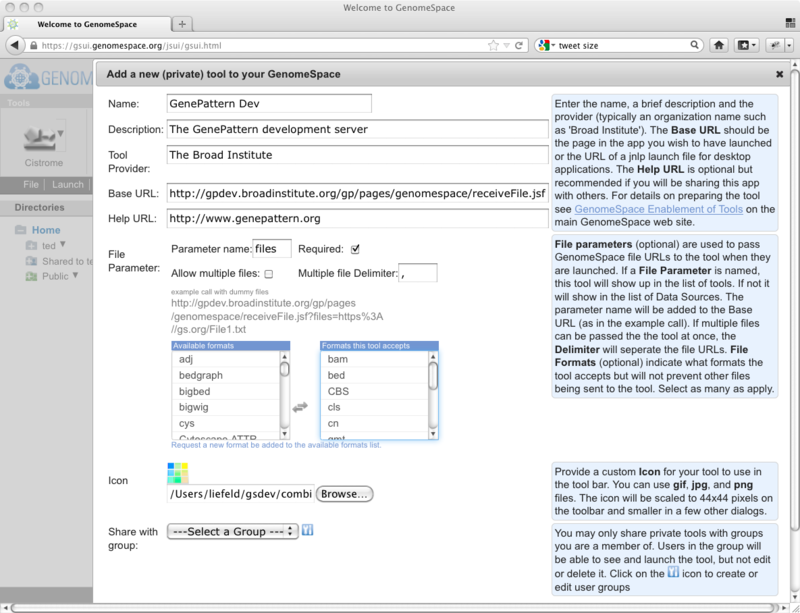
Help about what to use in each of the fields is available if you click the help icons (blue question marks) at the right side of the dialog. A completed tool information dialog (with the help showing) will look something like this:
That's it! Once your tool has been added, you should be able to launch it from GenomeSpace. Public Data: A differentiation map of hematopoiesis.We are pleased to announce that the data from the paper, Densely Interconnected Transcriptional Circuits Control Cell States in Human Hematopoiesis, Novershtern et al, Cell, Volume 144, Issue 2, 296-309, 2011, has been made available on GenomeSpace. You can access the data in GenomeSpace at the path /Public/SharedData/Datasets/DMap. AbstractThough many individual transcription factors are known to regulate hematopoietic differentiation, major aspects of the global architecture of hematopoiesis remain unknown. Here, we profiled gene expression in 38 distinct purified populations of human hematopoietic cells and used probabilistic models of gene expression and analysis of cis-elements in gene promoters to decipher the general organization of their regulatory circuitry. We identified modules of highly coexpressed genes, some of which are restricted to a single lineage but most of which are expressed at variable levels across multiple lineages. We found densely interconnected cis-regulatory circuits and a large number of transcription factors that are differentially expressed across hematopoietic states. These findings suggest a more complex regulatory system for hematopoiesis than previously assumed. |